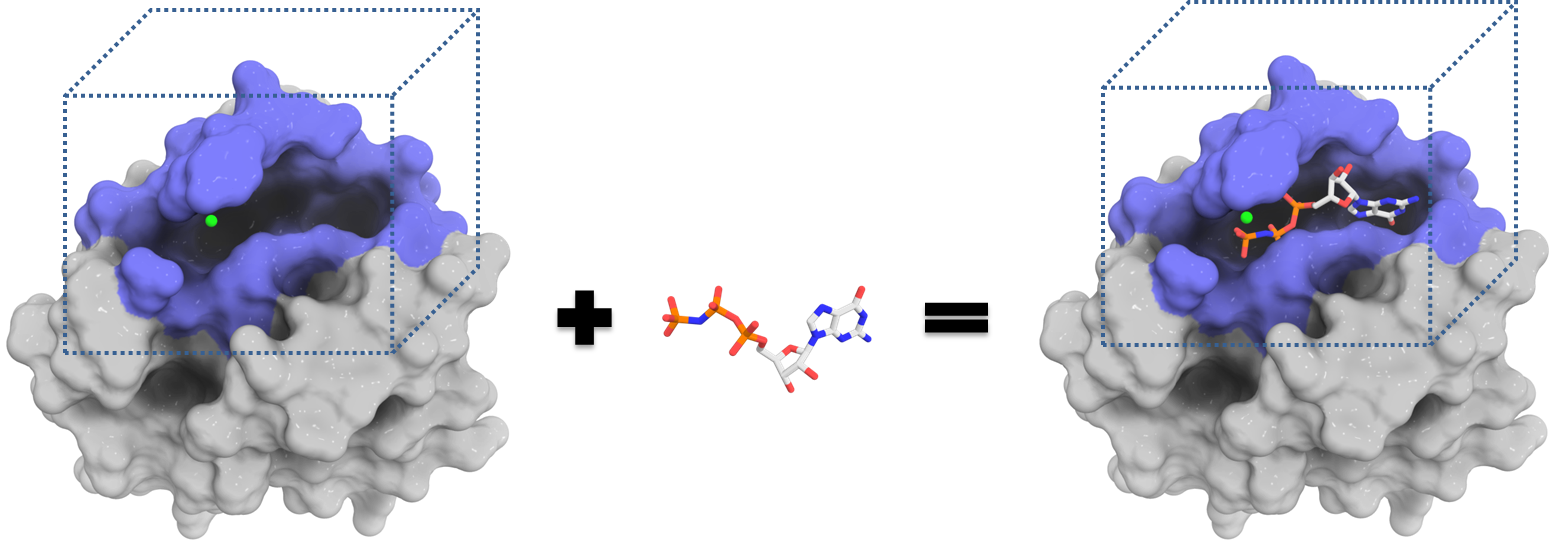
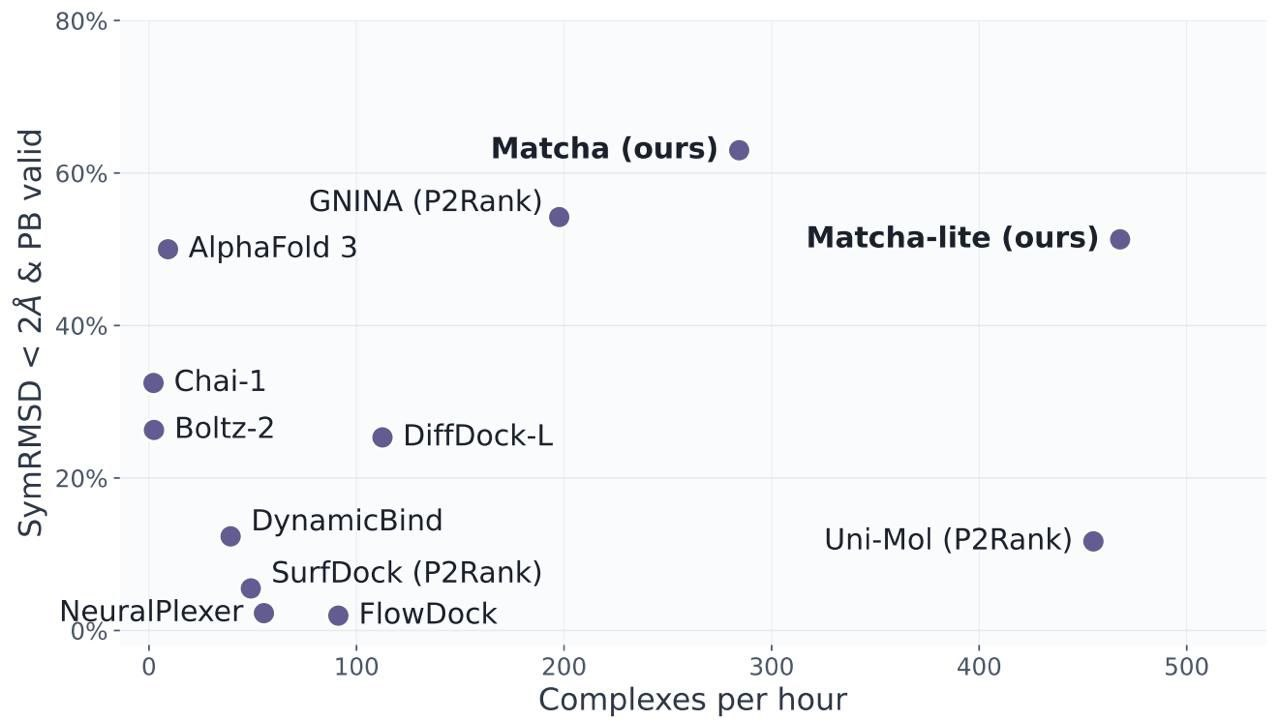
study
msc studies
phd studies
professional education
schedule
course catalog
academic calendar
independent studies period
industrial immersion
innovation workshop
admissions
about admissions
how to get into Skoltech (guide)
apply for msc programs
apply for phd programs
requirements and deadlines
internships
student community
student family support
organize an event at Skoltech
learn more